“交替极化”大概指的是下面这个现象?
这问题这么久没人答,大概是因为小众而且现在很少提了。把这个容易和诱导效应(Inductive Effect)放在一起讲,初学者可能会听懵。我也真见过有人不管有没有π键,一上来就说“这个基团有拉电子的诱导效应,推电子的共轭效应,所以综合看来是推电子的”。
现在很多书中把共轭效应(Mesomeric Effect or Resonance Effect )放到“离域化学键”体系中,并且主要强调的是共轭效应的稳定化作用。

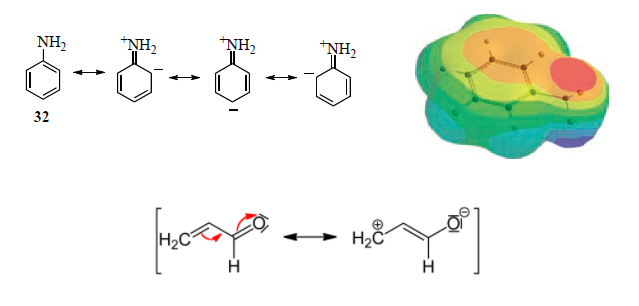
解释则多是用共振式 + 静电势能面图解释共轭效应,不怎么提交替极化(下图越红表示有较高的负电荷)。
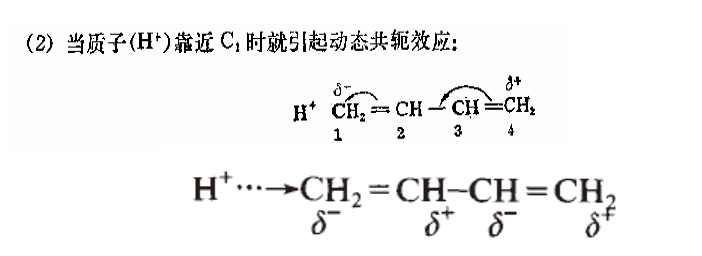
讲上面这些是要强调这个效应只出现在共轭体系里。为了说明这个理论,先来看诱导效应:
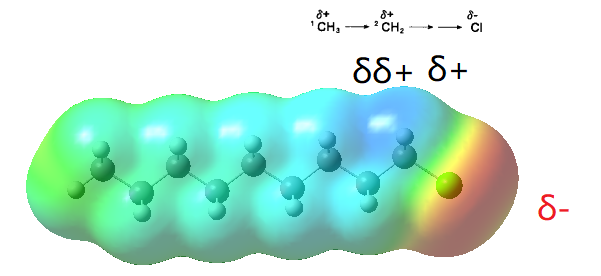
根据微扰理论的理解,像上面例子 R-Cl,吸电子的基团影响 C1 时, C1、C2、C3……的电荷必然要向 C1 偏移, 其结果是使 C1 呈δ+,因为σ键的电子比较“牢固”且“均匀”,这个影响的“符号”不会变化,而且随着距离增加会很快地衰减,总的影响是δ+、δ++、δ+++ 这样的。
总结思路就是在量子体系里用静电学的思路,讨论电子云密度在扰动后的变化。
用同样的思路讨论“离域化学键”体系,会发现π电子“不牢固”且“不均匀”。C2-C3 之间的电子云密度是比 C1-C2 和 C3-C4 低的。
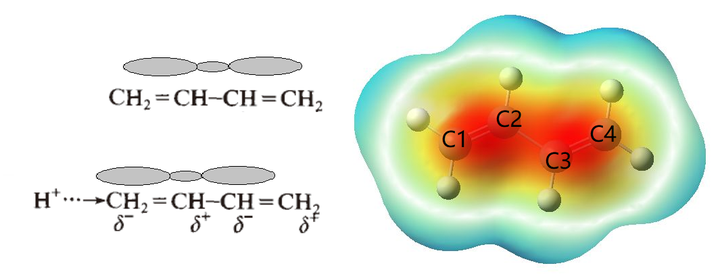
整体来看,当亲电试剂影响 C1 时,C1-C2 之间的电荷必然要向 C1 偏移,使 C1 呈δ-(指负电荷比微扰之前多,不是真的带负电荷),C2-C3 之间的电荷要向偏移,但由于 C2-C3 之间的电荷密度本来较低,所以对 C2 讲,移入它周围的电荷要少于它周围移出的电荷,因此呈δ+……这就是“交替极化”这个现象的一个解释[1]。总的影响是δ+、δ-、δ+、δ-……衰减慢是因为共轭π电子“离域”。
上面这个解释其实有很多问题,在量子体系里用静电学的思路思考问题不是很靠谱,而且只能解释链状共轭烃,环状共轭体系不适合用这个解释,因为苯环是键长平均化的体系。
所以现在不怎么讨论“交替极化”大概还因为它可以用Hückel 分子轨道理论里的交替碳氢化合物(alternant hydrocarbons,AH)规则代替。
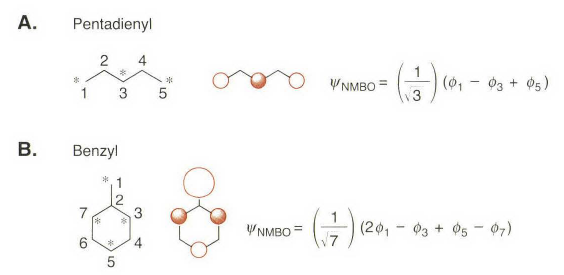
至于为什么共轭体系中π电子“不均匀”,有各种的解释。
共振论:
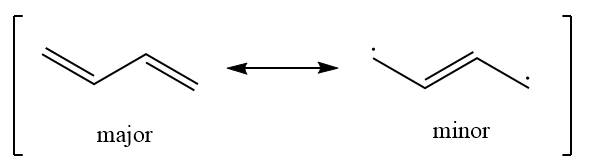
Hückel 分子轨道理论可以用“键级”解释。
MOT 之类的可以直接给出静电势能面。
我听过最深的解释大概是一维共轭体系中存在电子-声子相互作用(electron-phonon interaction),受布里渊区的影响,能带扭曲,键长和电子云密度都平均化的结构不是最稳定的。这个其实扯得有点远,我也只能记住上面几个关键词……有知道完整解释的可以说一说。
 微信扫一扫
微信扫一扫